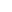FGF-21信号转导的分子层级工作机制
发表时间:2025-11-21FGF-21(成纤维细胞生长因子21)作为FGF家族内分泌亚群的核心成员,其信号机制呈现独特的共受体依赖性和代谢器官选择性。与旁分泌型FGF不同,FGF-21必须依赖β-Klotho作为共受体才能激活FGFR,形成"配体-共受体-受体"三元复合物。深入理解这一从蛋白构象变化到核转录调控的完整分子链条,对代谢性疾病研究和药物靶点验证实验设计具重要指导价值。
β-Klotho介导的共受体识别与特异性激活机制
FGF-21成熟肽段包含181个氨基酸,折叠成典型的β-三叶草结构。其核心特征在于C端延伸区(残基170-181),形成一段无规卷曲结构。这段延伸区直接结合β-Klotho的KL2结构域,亲和力Kd≈5×10??M。单独的FGF-21无法有效结合FGFR(Kd>10??M),必须预结合β-Klotho形成二元复合物,再由二元复合物识别FGFR。
β-Klotho是单次跨膜蛋白,胞外段包含两个KL结构域(KL1和KL2)。KL2与FGF-21结合后,构象变化传递至KL1,使KL1的C端环区与FGFR的D2-D3结构域距离缩短至3nm以内,满足FGFR二聚化的空间要求。这种"分子胶水"机制确保FGF-21仅作用于表达β-Klotho的组织(肝脏、脂肪、胰腺),实现信号的组织特异性。
实验中使用β-Klotho敲除小鼠原代肝细胞,FGF-21无法诱导ERK磷酸化,证明共受体不可或缺。某些商用FGF-21蛋白因氧化导致C端延伸区构象改变,β-Klotho结合力下降90%,表现为表观活性降低。质谱检测Met170氧化状态可作为质控指标。
FGFR1c-β-Klotho异源二聚体的空间组装与酪氨酸激酶激活
FGF-21-β-Klotho二元复合物优先结合FGFR1c亚型。FGFR1c胞外Ig样结构域D3的"酸性盒"序列(Asp316-Glu322)与β-Klotho KL1的正电荷表面形成静电互补。一个FGF-21-β-Klotho复合物同时交联两个FGFR1c分子,形成2:2:2化学计量比的三元复合物。
受体二聚化使胞内酪氨酸激酶结构域呈不对称排列,C端激酶段作为"激活子"磷酸化N端激酶段的激活环(A-loop)。FGFR1c的Tyr653和Tyr654位点磷酸化是激酶激活的分子开关。质谱分析显示,FGF-21刺激后10分钟,这两个位点磷酸化达到峰值,持续至2小时。
FGFR1c的Tyr766位点磷酸化招募PLCy1,启动PKC-NFAT通路。但FGF-21诱导的PLCy1激活强度仅为FGF-2的30%,这可能是FGF-21促有丝分裂活性较弱的原因。使用PLCy1抑制剂U73122,FGF-21的代谢调控功能仅下降15%,证明其糖脂代谢效应主要依赖其他通路。
ERK1/2通路的代谢转录调控网络
FGFR1c激活后,通过FRS2α支架蛋白招募GRB2-SOS复合物,启动Ras-MEK-ERK级联。FGF-21刺激下,ERK1/2磷酸化呈现双相动力学:初始峰值在15分钟,第二波在2-4小时出现。第一波由PLCγ-PKC路径快速启动,第二波依赖转录上调的MEK1蛋白合成。
磷酸化ERK1/2入核后,磷酸化转录因子Elk-1和STAT3。Elk-1结合脂肪酸氧化基因(ACADM、ACOX1)启动子,上调其表达。原代肝细胞中,FGF-21处理24小时使CPT1A mRNA提升8-10倍。这种效应在β-Klotho敲除细胞中完全消失。
ERK还磷酸化PPARα的Ser12位点,增强其转录活性。PPARα是FGF-21代谢效应的核心执行者。使用PPARα拮抗剂GW6471,FGF-21的降脂作用下降90%。ChIP-seq显示,FGF-21刺激后PPARα在基因组上的结合位点增加40%,其中70%位于代谢相关基因启动子区。
PI3K-AKT通路的协同作用与去磷酸化调控
FGFR1c同时磷酸化GAB1支架蛋白,招募PI3K的p85调节亚基。PI3K催化PIP2转化为PIP3,招募AKT至膜内侧。FGF-21刺激下,AKT的Ser473和Thr308位点磷酸化在30分钟达峰,持续至6小时。这种持续激活由PP2A磷酸酶调控。
AKT激活后磷酸化FOXO1的Ser256位点,促使其出核降解。FOXO1是糖异生关键基因G6PC和PCK1的转录激活因子。FGF-21处理使肝细胞FOXO1核定位下降80%,糖异生速率降低60%。这种效应在胰岛素抵抗模型中依然有效,证明FGF-21可作为胰岛素增敏剂。
AKT下游的mTORC1通路也被激活。FGF-21处理使S6K1磷酸化水平提升3倍,4E-BP1磷酸化提升4倍。但FGF-21主要激活mTORC1而非mTORC2。使用Raptor siRNA敲低,FGF-21的蛋白合成效应下降70%,而AKT-Ser473磷酸化不受影响,证明两条通路可分离调控。
PPARγ共激活与脂肪组织褐变机制
在脂肪组织中,FGF-21通过β-Klotho-FGFR1c复合物激活ERK,磷酸化PPARγ的Ser273位点。这个位点磷酸化不阻断PPARγ转录活性,反而招募PRDM16共激活因子,启动褐变基因程序(UCP1、CIDEA、DIO2)。白色脂肪细胞中,FGF-21处理7天使UCP1 mRNA上调50-100倍,氧耗率提升3-4倍。
这种褐变效应依赖共受体FGFR1c的表达。棕色脂肪细胞高表达FGFR1c,而白色脂肪细胞需经冷刺激或β肾上腺素激动剂上调FGFR1c后,才对FGF-21产生响应。使用FGFR1特异性抑制剂PD166866,FGF-21的产热效应完全消失。
FGF-21同时抑制脂肪细胞的脂质合成。通过ERK-PPARγ通路下调SREBP1c表达,使FASN和ACC1基因表达下降60-80%。这种抑制效应在ob/ob小鼠模型中改善脂肪肝,肝脏甘油三酯含量下降40%。
FGF-21的组织特异性清除与半衰期调控
FGF-21在循环中半衰期约1-2小时,主要清除器官是肾脏。肾小球滤过后,近端小管细胞表达的β-Klotho介导FGF-21重吸收和降解。这种清除机制导致FGF-21血药浓度波动大,需频繁给药。
PEG化修饰可延长半衰期至20-30小时。修饰位点选择N端Met1或Lys69对活性影响最小。Fc融合蛋白半衰期更长(>100小时),但二聚化可能改变受体激活模式。质谱分析显示,Fc-FGF-21更倾向于激活FGFR4而非FGFR1c,导致胆管增殖副作用。
蛋白降解酶DPP4在Val5-Pro6位点切割FGF-21,使其失活。使用DPP4抑制剂西格列汀可提升内源FGF-21水平30%。这种机制是GLP-1与FGF-21联用的理论基础。
实验研究中的浓度梯度设计与时间点选择
体外实验FGF-21有效浓度范围为1-1000 ng/mL。肝细胞糖生成抑制EC50约10 ng/mL,脂肪细胞褐变需100 ng/mL以上。浓度超过1000 ng/mL通常不提升效果,因受体已饱和。
时间动力学:ERK磷酸化在15-30分钟达峰,持续2小时。PPARα靶基因mRNA在2-4小时上调,蛋白表达在12-24小时。长期处理(>48小时)需每天换液补充FGF-21,维持有效浓度。
血清会结合FGF-21,降低游离浓度。10% FBS培养基中有效浓度下降30-50%。建议使用无血清或0.5% BSA培养基进行短期实验(<6小时),长期实验需相应提高FGF-21用量或每日补充。